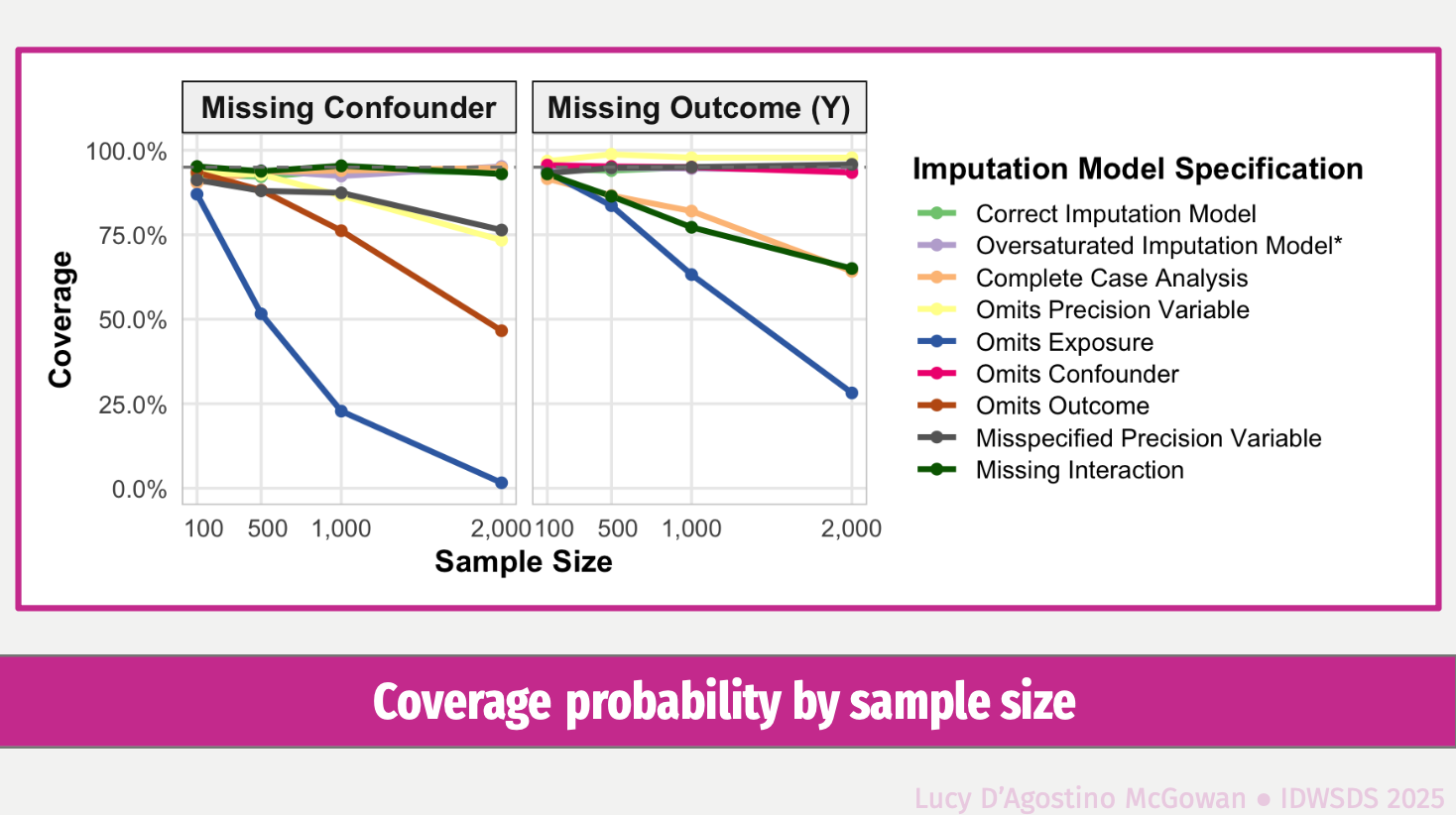
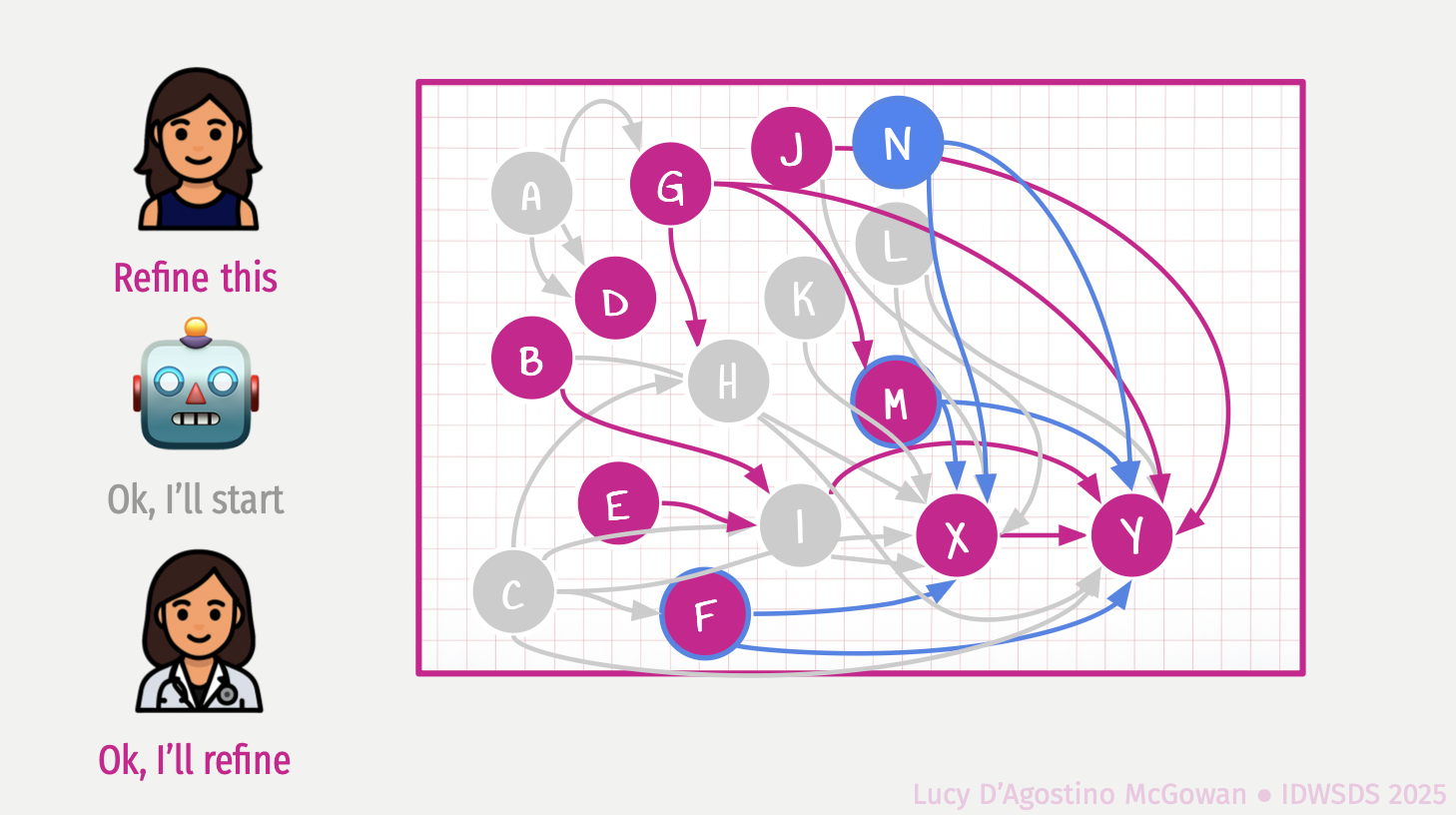
Mind the Gap: Causal Inference is Not Just a Statistics Problem
In this talk we will discuss some of the major challenges in causal inference, and why statistical tools alone cannot uncover the data-generating mechanism when attempting to answer causal questions. We will showcase the Causal Quartet, which consists of four datasets that have the same statistical properties, but different true causal effects due to different ways in which the data was generated. These examples illustrate the limitations of relying solely on statistical tools in data analyses and highlight the crucial role of domain-specific knowledge.